About AHP
(Acute Hepatic Porphyria)
AHP IS A RARE GENETIC
CONDITION THAT
CAN BE DEVASTATING1
EVERY AHP ATTACK MATTERS
AHP is characterized by acute, painful, potentially life-threatening attacks. It’s important to understand that your patient’s next AHP attack could have life-changing consequences.2
A single AHP attack can last 3 to 7 days and may require hospital admission and a prolonged inpatient treatment1,3

An AHP attack can cause extensive neurologic damage, some of which may be irreversible or take months to resolve2
Severe AHP attacks can progress to respiratory failure or paralysis, which could be fatal or lead to a temporary or permanent disability2,3
CLASSIFICATION OF PORPHYRIAS
Each type of porphyria involves a genetic defect in a heme biosynthesis pathway enzyme. They are classified as the following: acute hepatic porphyria (AHP)—which may present as a sudden neurovisceral acute attack—or photocutaneous porphyria.2,4,5
Using Major Clinical Manifestations for Classification of 9 Types of Porphyria5-7
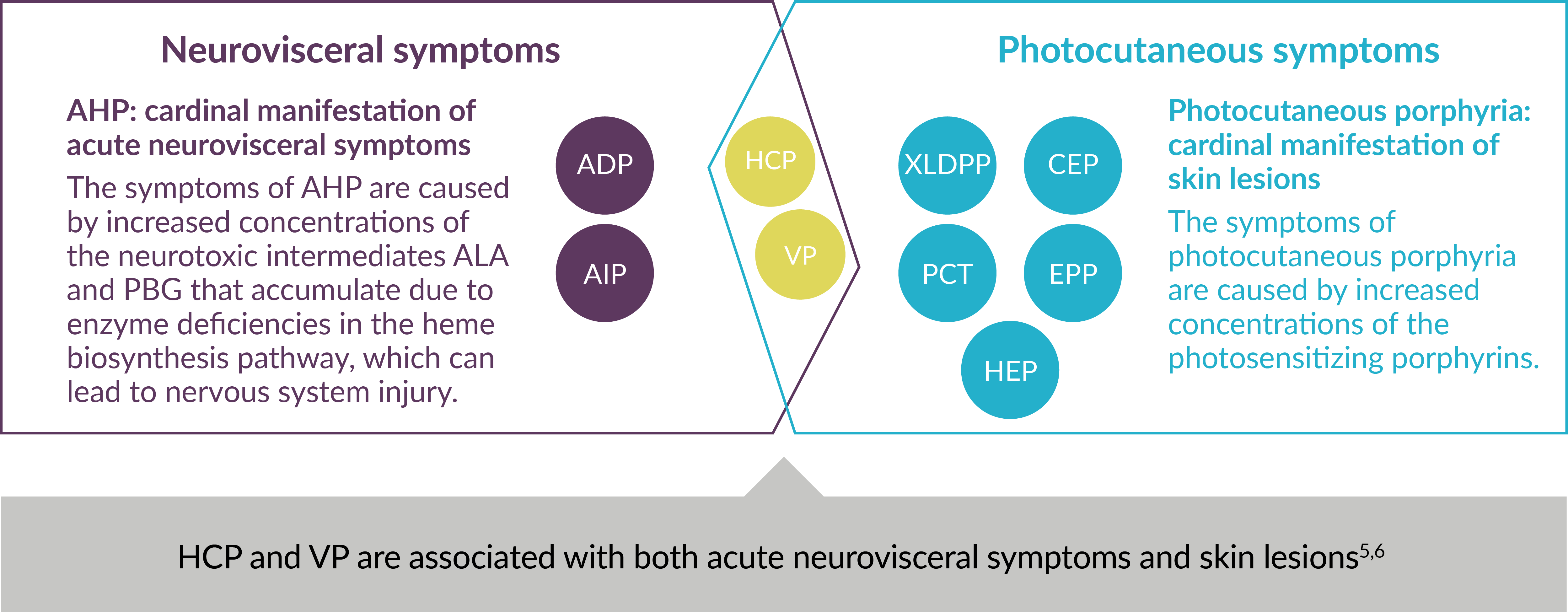
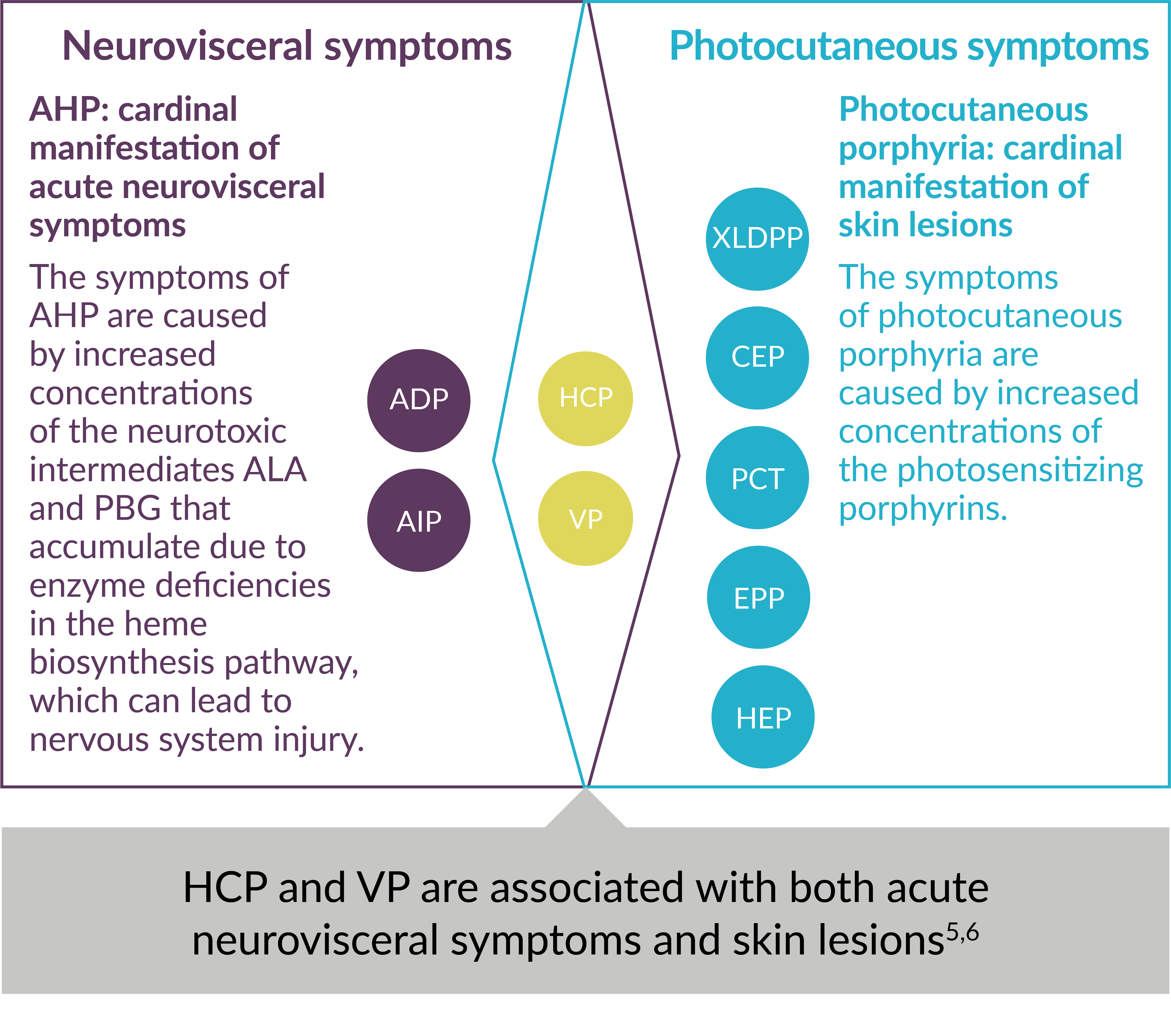
ADP=aminolevulinic acid dehydratase (ALAD)-deficiency porphyria; AIP=acute intermittent porphyria; ALA=aminolevulinic acid; CEP=congenital erythropoietic porphyria; EPP=erythropoietic protoporphyria; HCP=hereditary coproporphyria; HEP=hepatoerythropoietic porphyria; PBG=porphobilinogen; PCT=porphyria cutanea tarda; VP=variegate porphyria; XLDPP=X-linked dominant protoporphyria.
Causes of AHP
Porphyrias are genetic metabolic disorders of the heme biosynthesis pathway. AHP, a subset of porphyria, often presents as an acute attack that may require hospitalization. Attacks are triggered by an excess of neurotoxic porphyrin precursors, aminolevulinic acid (ALA) and porphobilinogen (PBG).1,4-6
Watch this Mechanism of Disease video to take a deeper look at the pathogenesis, signs and symptoms, and common methods used to help inform a diagnosis of AHP:
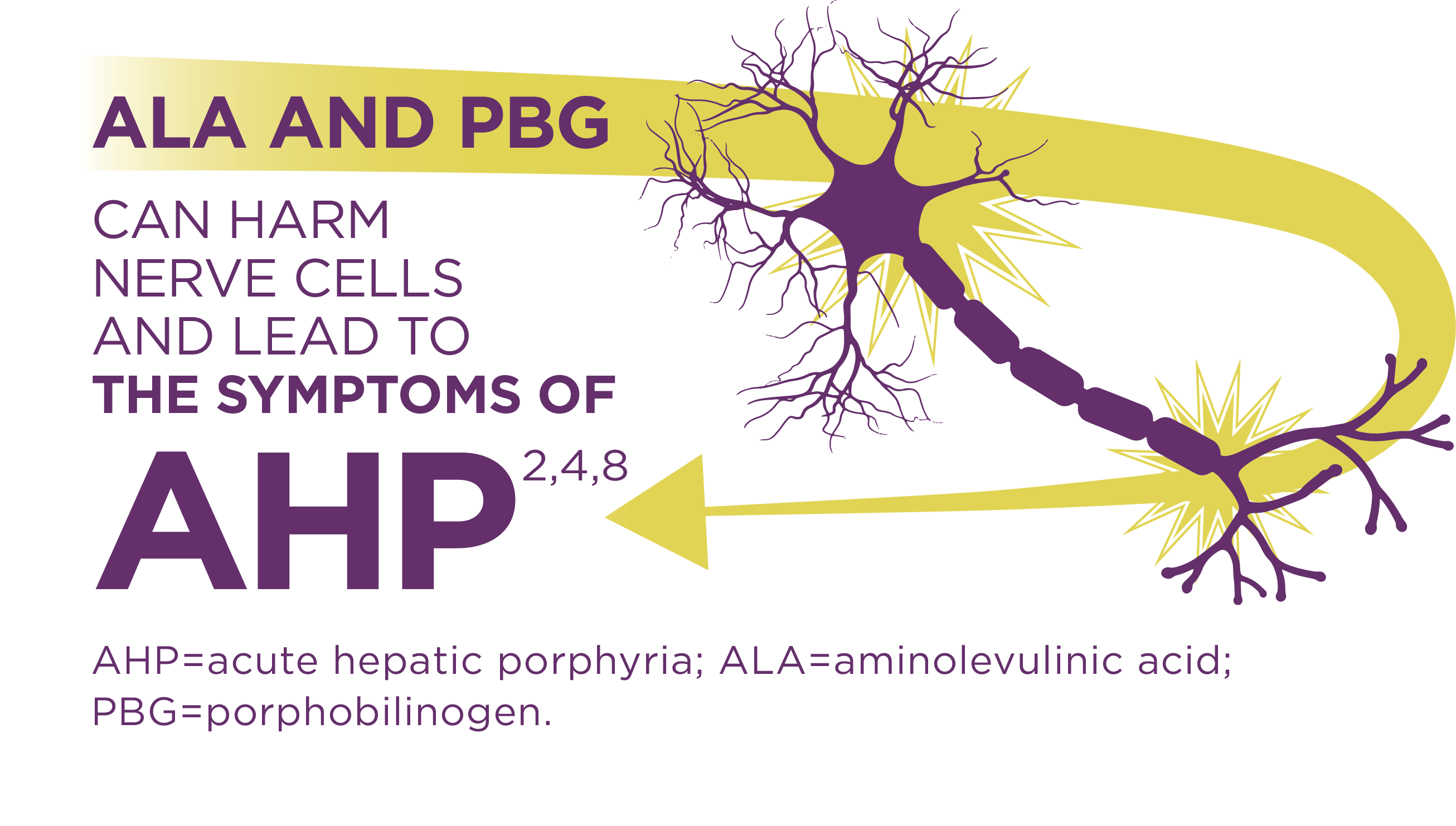

AHP is caused by enzymatic defects that result in accumulation in the liver of neurotoxic intermediates—aminolevulinic acid (ALA) and porphobilinogen (PBG). Upregulation of ALAS1 (aminolevulinate acid synthase 1) is a central pathophysiologic mechanism underlying AHP, and leads to this toxic accumulation. ALA and PBG then enter the circulatory system and are thought to cause neurologic lesions leading to dysfunction across the autonomic, central, and peripheral nervous systems.
Dysfunction in these systems can cause patients to have severe, diffuse abdominal pain plus one or more of the following symptoms: anxiety, confusion, nausea, vomiting, limb weakness and pain. This can potentially result in irreversible neuropathies and long-term disease complications such as kidney disease and hepatocellular carcinoma.
Elevated PBG is important as a highly specific marker that can help inform a diagnosis. ALA is believed to be the primary toxic intermediate responsible for causing acute attacks, ongoing symptoms between attacks, and prolonged or potentially permanent neuropathy in AHP. After an acute attack, the recovery period can be long and extend into residual chronic symptoms. Acute attacks generally last 3 to 7 days, but the recovery period can be long.1,2,4-6,8-11

Chronically elevated levels of ALA and/or PBG may present a higher risk of long-term complications such as chronic kidney disease, hepatocellular carcinoma, and hypertension3
Multiple studies have shown that symptomatic patients had 30 to 100 times higher risk of primary liver cancer vs reference controls12-15
AHP Consists of 4 TYPES
There are 4 types of AHP, which stem from different enzyme deficiencies in the heme biosynthesis pathway in the liver. About 80% of cases are acute intermittent porphyria (AIP), followed by variegate porphyria (VP), hereditary coproporphyria (HCP), and the extremely rare ALAD-deficiency porphyria (ADP). The prevalence of AIP may be underreported due to estimates based on patients with symptomatic disease only, rather than an enzyme mutation.1,4-6

Epidemiology of AHP
In the US, the overall prevalence of people diagnosed with symptomatic AHP is approximately 10 per 1 million people. It affects people of all races. The majority of cases occur in women of reproductive age, with most attacks occurring between the ages of 15 and 45. Importantly, the risk of long-term disease consequences, such as porphyria-related kidney disease or hepatocellular carcinoma, remains after menopause. Some patients experience debilitating and potentially life-threatening attacks.4-6,12,16-21
The majority of cases occur in women5,17,18*
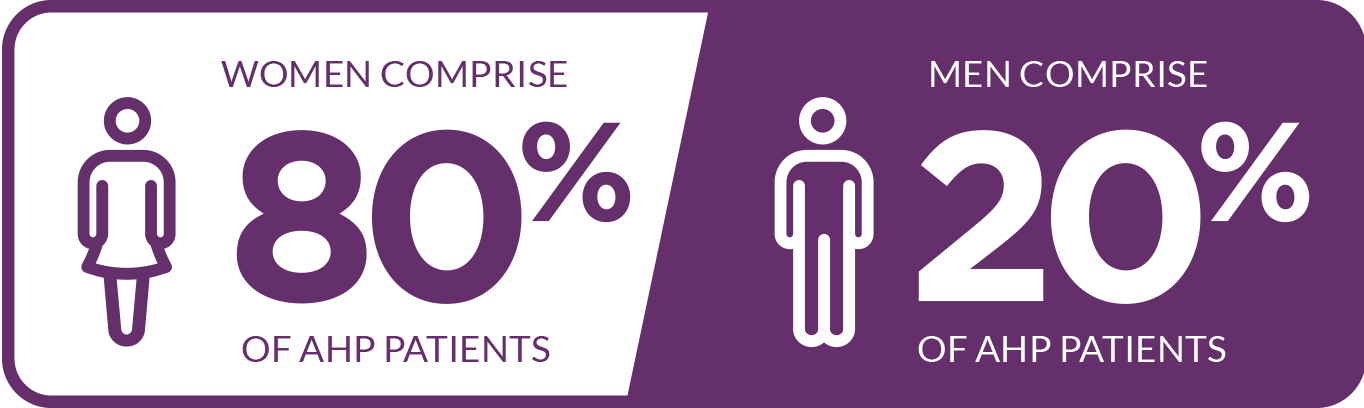

*In one study including 108 patients.
References: 1. Simon A, Pompilus F, Querbes W, et al. Patient. 2018;11(5):527-537. 2. Puy H, Gouya L, Deybach JC. Lancet. 2010;375(9718):924-937. 3. Neeleman RA, Wagenmakers MAEM, Koole-Lesuis RH, et al. J Inherit Metab Dis. 2018;41(5):809-817. 4. Anderson KE, Bloomer JR, Bonkovsky HL, et al. Ann Intern Med. 2005;142(6):439-450. 5. Bissell DM, Anderson KE, Bonkovsky HL. N Engl J Med. 2017;377(9):862-872. 6. Bissell DM, Wang B. J Clin Transl Hepatol. 2015;3(1):17-26. 7. Wang B, Rudnick S, Cengia B, Bonkovsky HL. Hepatol Commun. 2019;3(2):193-206. 8. Lin CS-Y, Lee M-J, Park SB, Kiernan MC. Clin Neurophysiol. 2011;122(12):2336-2344. 9. Szlendak U, Bykowska K, Lipniacka A. Adv Clin Exp Med. 2016;25(2):361-368. 10. Pischik E, Kauppinen R. Appl Clin Genet. 2015;8:201-214. 11. Balwani M, Wang B, Anderson KE, et al; Porphyrias Consortium of the Rare Diseases Clinical Research Network. Hepatology. 2017;66(4):1314-1322. 12. Baravelli CM, Sandberg S, Aarsand AK, Nilsen RM, Tollånes MC. J Intern Med. 2017;282(3):229-240. 13. Sardh E, Wahlin S, Björnstedt M, Harper P, Andersson DEH. J Inherit Metab Dis. 2013;36(6):1063-1071. 14. Andant C, Puy H, Bogard C, et al. J Hepatol. 2000;32(6):933-939. 15. Kauppinen R, Mustajoki P. Br J Cancer. 1988;57(1):117-120. 16. Elder G, Harper P, Badminton M, Sandberg S, Deybach J-C. J Inherit Metab Dis. 2013;36(5):849-857. 17. Ramanujam V-MS, Anderson KE. Curr Protoc Hum Genet. 2015;86:17.20.1-17.20.26. 18. Bonkovsky HL, Maddukuri VC, Yazici C, et al. Am J Med. 2014;127(12):1233-1241. 19. Ko JJ, Murray S, Merkel M, et al. Poster presented at: American College of Gastroenterology Annual Meeting; October 5-10, 2018; Philadelphia, PA. 20. Pallet N, Mami l, Schmitt C, et al. Kidney Int. 2015;88(2):386-395. 21. Ventura P, Cappellini MD, Biolcati G, Guida CC, Rocchi E; Gruppo Italiano Porfiria (GrIP). Eur J Intern Med. 2014;25(6):497-505. 22. Gouya L, Bloomer JR, Balwani M, et al. Presented at: 2017 International Congress on Porphyrins and Porphyrias; June 26, 2017; Bordeaux, France.